
Essais thérapeutiques et biais d’expertise. Le cas du « vaccin » de Pfizer
Le 10/10/2022 par Laurent Mucchielli

Dans le cadre des essais thérapeutiques, on ne mentionne pas assez l’existence d’un biais en amont essentiel, pour ne pas dire écrasant: le biais de financement. En aval, la façon dont les résultats sont présentés peut également donner lieu à un biais de notification. L’absence habituelle de la notification de la réduction du risque absolu par le traitement étudié (efficacité absolue) constitue en particulier un biais qui masque la juste appréciation du bénéfice de l’intervention thérapeutique, comme le montre la façon dont les résultats des essais cliniques réalisés pour le « vaccin » Comirnaty de Pfizer ont été rapportés par le fabricant, les autorités publiques et les médias
Par Michel CUCCHI, docteur en médecine, docteur en sociologie et cadre de la fonction publique, auteur de Influence & pandémies, expériences hasardeuses et tentations autoritaires (Éditions Marco Pietteur, 2022). L’auteur précise qu’il ne s’exprime pas au nom des institutions qui l’emploient et déclare son absence de conflit d’intérêts en relation avec les parties citées dans cette contribution
Introduction
Avec la manipulation du discours scientifique à laquelle se livre l’industrie du médicament depuis des décennies [10] et la complicité active des agences d’expertise en charge de son contrôle [13], il n’est désormais plus possible de se fier à leurs messages vantant leurs produits comme « sûrs et efficaces » puis de se plier aveuglément à leurs consignes. Le citoyen est en droit de mobiliser tout son esprit critique, de connaître la façon dont les produits destinés à l’administration humaine sont fabriqués par les industriels puis étudiés par les autorités sanitaires en vue de leur mise sur le marché. Ce droit universel dépasse de loin un « secret des affaires » qui ne protège que les intérêts d’une étroite minorité. Comme le déclarait le Comité consultatif national d’éthique pour les sciences de la vie et de la santé en octobre 1984, pour les soignants comme pour les gestionnaires, « il n’est pas conforme à l’éthique d’administrer un traitement dont on ne sait pas, alors qu’on pourrait le savoir, s’il est le meilleur des traitements disponibles, voire même s’il est efficace et s’il n’est pas nocif. L’évaluation d’un nouveau médicament est un devoir ». Pour les citoyens, il est devenu nécessaire de disposer des éléments permettant de juger de l’opportunité du recours aux produits proposés par l’industrie, de leurs bénéfices comme de leurs risques, avant de consentir aux soins de façon libre et éclairée.
Le présent article propose dans cette perspective d’explorer un domaine mal connu, celui du biais d’expertise hors du plan expérimental. Dans une première partie, nous allons voir que l’expression de l’efficacité d’une intervention thérapeutique devrait être formulée au travers de plusieurs indicateurs, notamment l’efficacité absolue et le nombre de sujets à traiter. Nous rappellerons à travers la notion de biais de financement que la présence de liens d’intérêt entre les expérimentateurs et l’industriel présentant son produit à l’expérimentation influe de façon majeure sur les résultats. Nous montrerons à travers le biais de notification que la façon dont les résultats parviennent à la population peuvent être la source d’une incompréhension et d’une mauvaise appréciation de l’intérêt du produit. Dans une seconde partie, nous illustrerons notre propos en reprenant la notification à la FDA des résultats de l’expérimentation du vaccin Comirnaty® de Pfizer sur la population de 18 à 74 ans en décembre 2020 et sa communication par les autorités sanitaires et les médias au cours de l’année 2021.
Essais thérapeutiques : indicateurs et biais
Une substance chimique porteuse d’une promesse thérapeutique ne devient médicament qu’après un protocole exigeant, l’essai thérapeutique, prenant de 7 à 12 ans entre l’idée d’un principe actif et sa commercialisation. Il comprend une période pré-clinique (ou de prérequis), consistant en des tests sur les animaux, préalable scientifique et éthique à toute administration chez l’homme, et une phase clinique composée de trois phases : la phase 1 est réalisée sur un petit nombre de volontaires, pour apprécier la tolérance (détermination de la dose maximale toxique). La phase 2, sur un effectif plus large, cherche à étudier l’efficacité pharmacologique et à ajuster la dose optimale pour la phase 3. Cette dernière est la phase des essais comparatifs sur de vastes effectifs. Elle se déroule normalement sur plusieurs années. Les résultats obtenus lors de la phase 3 permettent d’établir les rapports bénéfice-risque et coût-efficacité. L’autorité sanitaire compétente, la commission d’autorisation de mise sur le marché (AMM), vérifie la satisfaction des obligations scientifiques et réglementaires en vigueur en France et au sein de l’Union européenne et peut accorder une autorisation en cas de rapports bénéfice-risque et coût-efficacité favorables du point de vue des bénéficiaires potentiels du produit. En cas d’urgence, une autorisation peut être accordée avant la fin des études, en la conditionnant dans ce cas à la mise à disposition d’informations complémentaires (AMM conditionnelle). Ces autorisations permettent la prescription médicamenteuse dans une indication donnée. La phase 4, suivant l’AMM, n’est pas une situation d’essai. Elle consiste en une veille sur l’innocuité d’un produit, une précision des conditions d’utilisation et un ajustement de la posologie. La pharmacovigilance surveille les effets indésirables rares (que les phases précédentes n’ont pas permis de détecter) et en situation réelle.
De quelle efficacité parlons-nous ? Quelques définitions
Dans les essais cliniques de phase 3, il s’agit de réduire le risque de survenue d’un événement défavorable (ou morbide) choisi pour être représentatif de la maladie. L’expression de ce risque prend différentes formes. Le risque absolucorrespond au risque de survenue d’un événement en situation d’exposition (en l’occurrence, l’administration du produit expérimental), et le risque de référence correspond à ce même risque en situation de non-exposition. Le risque relatif (RR) est le rapport entre le risque absolu (au numérateur) et le risque de référence (au dénominateur). Il mesure la force de l’association entre l’exposition (l’administration du produit expérimental) et la survenue de l’événement défavorable. A groupes exposés/non exposés strictement comparables, la fréquence des événements dans les deux groupes est neutralisée : autrement dit, le risque relatif n’est pas sensible à la fréquence de l’événement dans la population.
L’efficacité relative (ER) d’un traitement exprime la réduction du risque relatif : ER = 1 – RR. Elle est en conséquence tout autant insensible aux situations cliniques ou épidémiologiques : c’est la seule efficacité du produit expérimental qui est ainsi mesurée. Cette grandeur permet pour l’essentiel la comparaison des protocoles thérapeutiques ou des produits expérimentaux entre eux, indépendamment de la situation sanitaire et de la population étudiée. Elle ne permet pas de fonder un besoin d’intervention. L’exposition d’une efficacité relative couvre ainsi une hypothèse implicite, la certitude de l’opportunité de l’intervention médicamenteuse, qu’il serait pourtant important de questionner en préalable.
L’efficacité absolue (EA) d’un traitement exprime une réduction du risque absolu (RRA) sous la forme d’une différence : EA = (risque absolu) – (risque de référence). Elle intègre donc le risque de référence et permet d’établir le bénéfice attendu pour le patient de la prise du produit expérimental en rapport avec le critère représentatif de la maladie (c’est le « bénéfice » du rapport bénéfice-risque). Enfin, le nombre de sujets à traiter (NST) est l’inverse de l’efficacité absolue : NST = 1/EA. Il exprime le nombre nécessaire de patients à traiter pour éviter un événement donné. C’est une information essentielle pour le prescripteur ou le gestionnaire : il représente l’effort de prise en charge pour prévenir un événement.
Une notification correcte des résultats doit ainsi compter au moins trois grandeurs complémentaires : (1) l’efficacité absolue, qui permet d’emblée d’estimer un bénéfice pour la personne « bénéficiaire » du traitement, (2) son inverse, le nombre de sujets à traiter, qui permet d’estimer un effort de prise en charge, enfin (3) l’efficacité relative, dans l’hypothèse où plusieurs approches thérapeutiques sont envisageables.
Biais d’expertise d’origine extra-méthodologique
Un biais d’expertise peut être défini comme une erreur systématique contenue dans l’information communiquée aux décideurs, aux professionnels ou à la population. Dans un chapitre du manuel Essais thérapeutiques, mode d’emploi intitulé « Qu’est-ce qu’un biais ? » [9, pp. 139-141], Françoise Doyon (INSERM, Unité 287, Épidémiologie des cancers, Villejuif) explique que « le terme de biais est utilisé pour désigner toute différence systématique entre les groupes que l’on compare dans un essai thérapeutique, hormis les traitements que l’on étudie ». Une entorse à la comparabilité des groupes, structurelle ou progressive (avec notamment l’important problème des sujets exclus ou perdus de vue) constitue effectivement une source importante de biais. Mais il en existe bien d’autres, qui ne relèvent pas de la méthode expérimentale mais plutôt de l’organisation de l’expertise et plus généralement de la manipulation de l’autorité scientifique [7, p. 69sqq.]. Ces sources de biais apparaissent soigneusement détachées du champ de l’essai thérapeutique et se dérobent ainsi au regard des professionnels de santé et de la population, destinataires finaux de ces informations [13]. Nous examinerons dans cette partie deux sources bien établies de biais d’expertise d’origine extra-méthodologique : le biais de financement (en amont du plan expérimental) et le biais de notification des résultats (en aval de celui-ci).
Le biais de financement (funding effect)
L’industrie du médicament dispose de ressources financières considérables qui lui permettent de contrôler la production scientifique. Le biais de financement est l’un de ces leviers de contrôle.Formalisé au milieu des années 1980 par Richard Davidson, il établit que les travaux sponsorisés par les industriels diffèrent dans leur résultat de ceux financés par d’autres sources [8]. Ce biais est particulièrement dangereux dans les secteurs de l’alimentation ou du médicament, où les agences sanitaires demandent aux industriels de financer l’essentiel des études de toxicité conduisant à la mise sur le marché de leurs produits.
Depuis les années 1990, des médecins et des sociologues étudient ce phénomène en réexaminant les résultats des travaux et les orientations des disciplines en fonction des financements perçus. Le Dr Ralph Walton, un psychiatre, a par exemple établi en novembre 1996 que sur les 164 publications relatives à la sécurité de l’aspartame dans les revues à comité de lecture, 74 avaient été financées par « l’industrie de l’aspartame » : toutes ces 74 publications concluaient que l’aspartame était sans danger, et toutes les publications non financées par l’industrie « concluaient qu’il y avait un problème avec la substance », soit une corrélation à 100% entre le financement des études et leur résultat : « tragiquement, l’argent est très puissant », conclut le Dr Walton [19]. En 1998, les Tobacco documents ont permis de montrer que les résultats des études sur le tabagisme passif différaient selon un facteur explicatif principal, l’affiliation de l’un des auteurs à l’industrie du tabac [2]. Les preuves de l’existence du phénomène s’accumulent ainsi dans la plupart des domaines industriels : le médicament bien sûr [15], mais aussi le sucre [14], les biotechnologies [11], les pesticides [4], la pétrochimie [20]. Enfin, l’équipe du Professeur Raoult parvient à montrer la relation entre les liens d’intérêt des signataires des études et leur résultat concernant l’efficacité du remdésivir et de la chloroquine [16] ainsi que de l’hydroxychloroquine [17] sur le covid. Ainsi, par construction, les essais contrôlés par l’industrie, qui sont censés éviter les biais, se caractérisent d’abord par leur aveuglement au premier d’entre eux, celui de leur financement. C’est la tâche aveugle de ces essais volontiers présentés comme le sommet de la preuve scientifique.
Le biais de notification des résultats (outcome reporting bias)
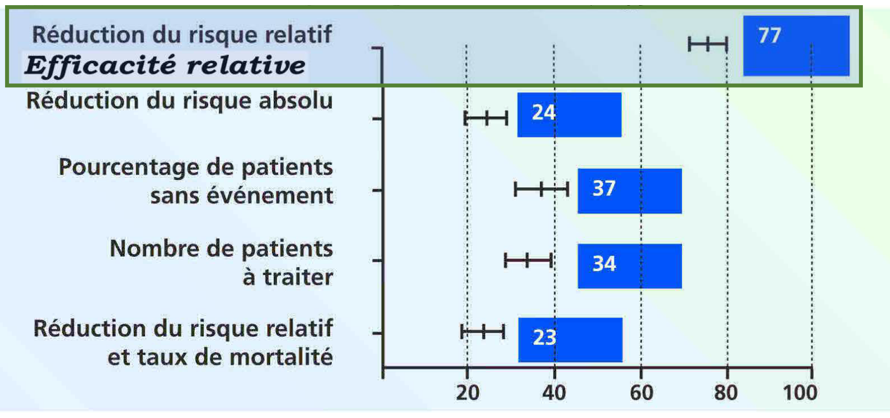
L’équipe de Marco Bobbio a étudié en 1994 l’impact de la présentation des résultats sur la décision thérapeutique [3]. Les résultats de l’étude Helsinki sur l’effet d’un médicament hypocholestérolémiant sur le risque d’AVC à 5 ans de la survenue d’un infarctus du myocarde ont été rapportés selon plusieurs indicateurs présentés comme extraits d’essais cliniques différents : ER = 34% ; EA = 1,41% ; Pourcentage de patients sans événement : 97,3% vs 95,9% sous placébo ; NST = 71. Il en résulte que l’expression en termes de réduction du risque relatif (efficacité relative) a l’effet prescripteur de loin le plus puissant (77/100). Les autres expressions ont des effets prescripteurs proches (37, 34, 24, 23). Ainsi, la communication de la seule efficacité relative influence abusivement les médecins et leurs patients dans le sens de l’interventionnisme médicamenteux ou médical. De nombreuses publications ont mis par la suite cet abus en évidence, en particulier pour la communication des résultats des vaccins à ARNm [5].
Tenant compte de ce biais de notification, la FDA recommande dans son « Manuel de communication des risques et bénéfices » de 2011 [6, p. 60] de communiquer l’efficacité absolue : « Fournissez des risques absolus, pas seulement des risques relatifs. Les patients sont indûment influencés lorsque les informations sur les risques sont présentées en utilisant une approche par le risque relatif ; cela peut entraîner des décisions sous-optimales » (en gras dans le texte). Mais la FDA ajoute à cette consigne une phrase au conditionnel qui peut signifier qu’elle fermera les yeux si, donc, les patients sont influencés de façon abusive, au risque de prises de décision dégradées : « Ainsi, un format de risque absolu devrait être utilisé ».
Examen et communication des données produites par l’industrie
Pour illustrer ces éléments avec un exemple concret, nous nous intéressons dans cette partie à la communication des résultats de l’essai clinique de Pfizer sur la population de 18 à 74 ans à la FDA en décembre 2020 et à leur notification à la population. Les données correspondantes sont en effet les premières à être largement disponibles, depuis la requête FOIA (Freedom of Information Act) du cabinet d’avocats new-yorkais Siri & Glimstad : malgré ses engagements de transparence, la FDA étasunienne a dû en effet être poursuivie en justice en septembre 2021 pour rendre publiques les données de Pfizer sur lesquelles elle s’est fondée pour lui accorder l’autorisation conditionnelle (la FDA réclamait pour sa part un délai de 55 ans avant publication). Ce sont également les plus détaillées. Le produit correspondant est également le premier autorisé, le plus injecté, à des âges toujours plus étendus (des personnes âgées aux adolescents puis les enfants), y compris chez les femmes enceintes et allaitantes. La phase 3 de l’essai multicentrique randomisé en aveugle réalisé par Pfizer pour le vaccin à ARN messager Comirnaty® (BNT162b2) a donné lieu à la publication d’une documentation en date du 10 décembre 2020 pour la population de 18 à 74 ans, du 9 avril 2021 pour celle de 12 à 15 ans et du 26 octobre 2021 pour celle de 5 à 11 ans.
Les résultats des essais ayant visiblement fait l’objet d’une communication à caractère quasi-publicitaire, nous prenons le parti de mêler ces résultats avec leur communication par les médias. Par ailleurs, nous nous prêtons à l’exercice de prendre ces données au sérieux : nous n’exprimons pas d’interrogation sur leur qualité, puisqu’elles ont servi de base scientifique à l’autorisation conditionnelle de mise sur le marché du « vaccin » expérimental.
Études d’efficacité
Pour la phase 3 de l’essai, les 40 000 personnes ont été divisées en deux groupes comparables (par l’âge, les comorbidités, etc.) d’approximativement 20 000 personnes, l’un destinée à recevoir le produit de Pfizer, l’autre un placébo (sérum physiologique).
Efficacité de l’évitement du covid symptomatique
L’objectif est d’évaluer l’efficacité du BNT162b2 (la solution expérimentale commercialisée sous le nom de Comirnaty®) avec comme critère principal la prévention du développement du covid symptomatique à partir de 7 jours après la 2e dose de vaccin chez les participants sans antécédent d’infection Covid-19. Le covid symptomatique (personne « malade ») est défini comme (1) un test PCR positif et (2) au moins l’un des symptômes suivants : fièvre, toux, essoufflement, frissons, douleurs musculaires, anosmie, agueusie, mal de gorge, diarrhée, vomissement. Les données d’efficacité vont de 19 à 42 jours.
Après la levée de leur anonymat, il apparaît que sur les 170 « malades », 162 ont reçu le placebo, et 8 le vaccin, ce qui conduit à une efficacité relative de 95% et une efficacité absolue de 0,84%.
Ce qu’auraient dû dire les autorités sanitaires pour notifier le bénéfice attendu pour la population, c’est que selon les données de Pfizer de décembre 2020, 99,12% des personnes non vaccinées n’ont pas eu le covid, contre 99,96% des vaccinés : dans le cadre de l’essai clinique de Pfizer, la « vaccination » diminuait pour chacun son risque de contracter un covid symptomatique en moyenne de 0,84%.
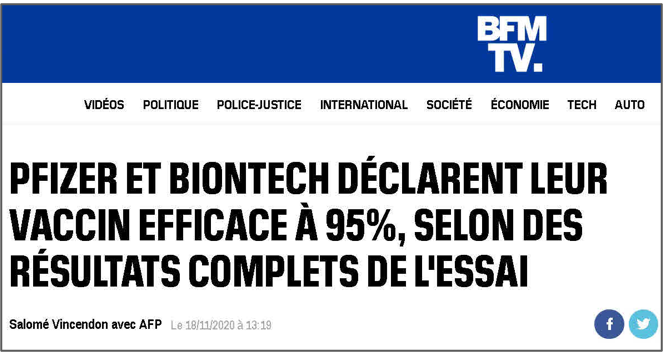
Les chiffres communiqués dans les médias mainstream par exemple ici BFMTV) sont très éloignés de cette évaluation au plus juste du bénéfice pour la population. Les enseignes Pfizer et BioNTech imposent une efficacité établie à 95% dès le 18 novembre 2020, avant même la notification de leurs résultats à la FDA à partir de résultats présentés comme « complets ». Ce chiffre de 95% correspond exactement à ce qui ressemble fort à une prophétie exprimée par Bill Gates dans sa communication sur la « première pandémie moderne », le 23 avril 2020 : « Si au printemps 2021, les gens vont à de grands événements publics – comme un match ou un concert dans un stade – ce sera parce que nous avons un traitement miraculeux qui a donné aux gens la confiance nécessaire pour sortir à nouveau. Il est difficile de savoir précisément quel est le seuil, mais je soupçonne qu’il est de l’ordre de 95% ; c’est-à-dire que nous avons besoin d’un traitement efficace à 95% pour que les gens se sentent en sécurité dans les grands rassemblements publics. Bien qu’il soit possible qu’une combinaison de traitements ait une efficacité supérieure à 95%, ce n’est pas probable, nous ne pouvons donc pas compter dessus. Si nos meilleurs traitements réduisent le nombre de décès de moins de 95%, nous aurons toujours besoin d’un vaccin avant de pouvoir revenir à la normale. » Le produit de Pfizer a ainsi tous les atours du « traitement miraculeux » vanté par Bill Gates.

Dans les mois qui suivent, ce chiffre est martelé, voire dépassé, en s’éloignant bien loin de la signification d’une efficacité relative, comme dans cette dépêche d’Associated Press du 29 juin 2021 (Figure 4). Selon Andy Slavitt, un ancien conseiller de l’administration Biden, « 98% à 99% des Etasuniens qui meurent du coronavirus ne sont pas vaccinés ». Rochelle Walensky, la directrice des CDC, estime le vaccin est si efficace que « presque chaque décès dû au covid, en particulier chez les adultes, est désormais entièrement évitable », ce qui rendrait ces décès « particulièrement tragiques ». Le produit se révélerait plus efficace encore que dans les essais, non plus pour éviter des formes symptomatiques mais directement des décès.
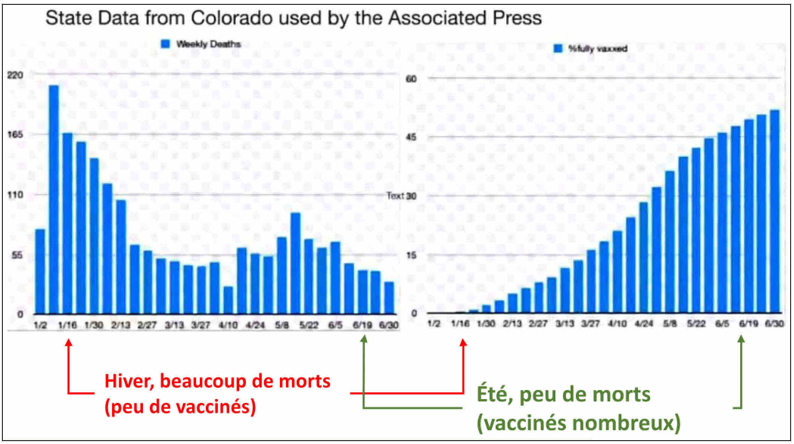
La dépêche évoque des « données gouvernementales » pour étayer ces pourcentages, mais elles ne sont disponibles ni à l’agence Associated Press, ni aux CDC. Le rédacteur scientifique en chef du journal en ligne The Defender, le Dr Madhava Setty, a retrouvé ces données : elles ne sont pas issues d’une étude traditionnelle (un groupe de personnes « vaccinées », un groupe « non vaccinées » idéalement comparables), ce que le lecteur pourrait comprendre en l’absence d’information particulière, mais de l’État du Colorado. L’étude prend en compte l’ensemble des décès sur la période du 1er janvier à la fin de juin 2021. Elle mélange deux époques bien différentes, celle du début du semestre, en hiver, avec des décès nombreux (et peu de personnes vaccinées), et celle de sa fin, en été, avec peu de décès (et un nombre élevé de personnes « vaccinées »). Outre l’absence des données sources, la tromperie réside notamment dans l’assimilation de l’ensemble de la période à la situation du moment où sort la dépêche, à la fin juin (le chapeau de la dépêche précise de façon trompeuse que « presque tous les décès du covid aux États-Unis surviennent maintenant chez des personnes qui n’étaient pas vaccinées, une démonstration stupéfiante de l’efficacité des injections »).
Efficacité de l’évitement d’une forme grave de covid
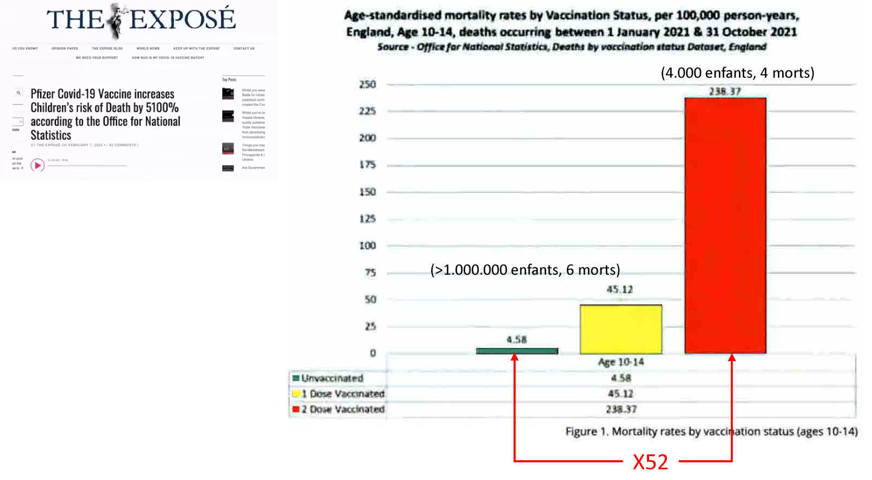
Avant de traiter de l’efficacité du traitement sur les formes graves, considérons cette publication de février 2022 du journal britannique en ligne The Exposé qui annonce dans un titre-choc que « le vaccin anticovid de Pfizer augmente le risque de mort infantile de 5100%, selon l’Office de la statistique britannique » (Figure 5). Cette proportion de décès du fait d’une « vaccination » par deux injections apparaît d’emblée invraisemblable et suspecte. En examinantles données, le déséquilibre des groupes apparaît manifeste (plus d’un million d’enfants non « vaccinés » contre 4000 enfants « vaccinés » avec deux doses) mais aussi le nombre de décès trop faible (10 morts : 6 dans le groupe des « non vaccinés », 4 dans le groupe des « vaccinés ») : l’influence d’un seul décès sur le résultat apparaît démesuré, et la zone d’incertitude trop grande pour que cette différence ait la moindre chance d’apparaître significative.
Les mêmes interrogations sur la qualité des données peuvent être soulevées avec l’étude de l’efficacité du produit de Pfizer sur les formes graves du covid, qui entraînent une hospitalisation, des anomalies biologiques le plus souvent non monitorées ou encore un décès. Cette étude donne lieu à 5 tableaux d’occurrence différents (Tables 16 à 20, pp. 65 à 69). Seul le tableau n°18 émet un signal susceptible d’être pris en considération, les autres tableaux affichent un intervalle de confiance encadrant largement la valeur zéro : c’est en effet le seul qui prend en compte le développement d’une forme grave de covid de la première dose à la fin de la période de surveillance, les autres se limitant aux effets secondaires à l’issue de la seconde dose. Un même nombre d’événements que dans l’article du journal en ligne (10 au total) sert de base à l’étude de l’efficacité du produit de Pfizer sur les formes graves : 6 événements après la première dose, 4 événements après la seconde dose. Compte tenu du très faible nombre de formes graves, l’interprétation objective des données est qu’aucune efficacité n’est démontrée sur les formes graves par manque de cas. Dans sa recommandation du 23 décembre 2020 (p. 6), la Haute autorité de santé exprime cependant un avis différent (« A ce stade, les données ne permettent pas de confirmer l’impact de la vaccination par le BNT162b2 sur les hospitalisations, les hospitalisations en unité de soins intensifs, ni de démontrer un impact sur la mortalité. Il est toutefois noté un effet sur l’incidence des formes sévères »), et les plus hautes autorités de l’État s’empressent alors de communiquer sur cette efficacité alléguée.
Nous sommes ainsi contraints de prendre au sérieux ce que montrent les indicateurs calculés à partir de ces données : sur cette base de 10 événements, l’efficacité relative calculée est de 88,9% (arrondie à 90% dans la communication au public), et l’efficacité absolue de 0,04%.
Ce qu’auraient dû dire les autorités sanitaires pour communiquer le bénéfice attendu pour la population, c’est que, pour autant que les données de Pfizer de décembre 2020 soient reconnues comme significatives, 99,958% des personnes non vaccinées n’ont pas eu de covid sévère, contre 99,996% des vaccinés : dans le cadre de l’essai clinique de Pfizer, la « vaccination » diminuait pour chacun son risque de contracter un covid sévère en moyenne de 0,04%.

Là encore, seule l’interprétation triomphante parvient à percer le mur de la communication mainstream, avec dans cette dépêche de l’AFP du 6 mai 2021 qui se veut « factuelle » (Figure 6) la stigmatisation des personnes osant des interprétations différentes (ici, un « pharmacien hospitalier de Cholet »).
Au total : une communication biaisée, des citoyens abusés
Bien que ne manifestant aucune considération à l’égard du biais structurel de financement que constitue la conduite des essais thérapeutiques par les industriels de leur propre produit, la FDA leur recommande cependant de communiquer l’efficacité absolue de leur produit, et pas uniquement leur efficacité relative, car cela influence abusivement les patients et les médecins. Mais elle fait en sorte en même temps de signifier que si les industriels ne le font pas, elle est prête à fermer les yeux sur la consigne, une attitude suivie avec constance depuis que la recommandation a été émise. En conséquence, les industriels du médicament n’en font rien. Même la publication de Polack et al. [18], faisant référence en matière de communication des essais cliniques de Pfizer, ne fait pas mention de l’efficacité absolue. La communication institutionnelle relaie les messages des industriels, alors qu’elle pourrait d’elle-même ajouter les informations manquantes ou la corriger pour ne pas induire la population en erreur. Les médias mainstream se placent d’eux-mêmes en porte-voix de cette propagande et ne permettent pas à d’autres sources d’information d’être entendues.
Dans le cas de la vaccination anti-covid, en ne communiquant pas l’efficacité absolue, c’est-à-dire la réduction du risque de référence par la vaccination (inférieur à 1% dans laFigure 7), l’estimation du bénéfice pour le citoyen demeure masquée, et celui-ci n’a pas les ressources pour l’établir correctement.Abusivement influencé, il se trouve conduit voire contraint de prendre des mauvaises décisions pour lui-même et ses proches, dans la conduite de sa vie sociale comme dans son environnement professionnel. La persistance d’un tel biais au bénéfice des industriels est incompatible avec le recueil d’un consentement éclairé et met en doute l’intégrité de l’expertise médicamenteuse dans son entièreté.
Effets indésirables et calcul du rapport bénéfice-risque
Les effets indésirables liés aux vaccins rendus publics sont fréquents et le plus souvent bénins : douleur locale (86%), fatigue (63%), maux de tête (55%), douleurs musculaires (38%). Les réactions de niveau 3, empêchant une vie quotidienne normale, toucheraient 4,6% des volontaires. Ces informations lénifiantes sont les seules à être reprises en substance par la presse [12]. Les données de Pfizer de décembre 2020 montrent pourtant un risque d’effets indésirables graves (EIG) associé à la prise du produit susceptibles d’entraîner une hospitalisation, des anomalies biologiques non explorées ou encore un décès.
Le Tableau n°8 fait apparaître le chiffre de 126 EIG (Any serious adverse event) sur un effectif de 21 621 personnes vaccinées, soit un pourcentage de 0,583% personnes « vaccinées » (arrondi à 0,6%) affectées par au moins un EIG. L’essai de Pfizer fait également état d’un effectif de 21 631 personnes ayant reçu un placebo (sérum physiologique) par lequel 111 EIG sont survenus, soit un pourcentage de 0,513% (arrondi à 0,51%) un chiffre étonnamment élevé pour une population adulte en bonne santé sous sérum physiologique.
Nombre de personnes à « vacciner »
Le nombre de sujets à « vacciner » pour éviter l’événement correspondant au critère principal est obtenu par l’inverse de l’efficacité absolue du produit. Ainsi :
- pour éviter un covid symptomatique, il faut « vacciner » 1 / 0,0084 personnes, soit 119,04 personnes (arrondi à 120 personnes), soit 120 injections pour une dose, 240 pour deux doses, etc.
- pour éviter une hospitalisation pour covid sévère, il faut « vacciner » : (9-1) / 21686 = 0,0003689016 (0,04%) = 8 / 21686 = 1 / 2710,75, soit 2 711 personnes (calcul rapide : 1 / 0,0004 personnes, soit 2500 injections pour une dose, 5000 injections pour deux doses, etc.)
Le nombre de sujets à vacciner pour éviter une forme grave de covid constitue une information capitale pour mesurer l’engagement soignant. Cette information n’est jamais apparue, ni dans l’interprétation des résultats de Pfizer, ni à notre connaissance dans les interprétations des différentes agences sanitaires en charge de fournir des recommandations à l’autorité publique ou dans les médias.
Vaccination en population générale : un rapport bénéfice-risque défavorable
Nous avons vu plus haut que (1) pour éviter une hospitalisation pour covid sévère, le nombre de personnes à vacciner est de 2710,75 (arrondi à 2711) et (2) 0,583% personnes « vaccinées » (arrondi à 0,6%) affectées par au moins un EIG lié au « vaccin », c’est-à-dire conduisant soit à une hospitalisation, soit à un événement indésirable grave n’entraînant pas d’hospitalisation (comme la mort au domicile par exemple). Dans une population de 18 à 74 ans en bonne santé (voire en meilleure santé que la population générale), l’essai de Pfizer révèle qu’il convient alors d’attendre 2710,75 x 0,583% = 15,80 (16 EIG) (calcul rapide : 2 500 x 0,6 = 15 EIG). En tenant compte du groupe placébo, au nombre d’EIG étonnamment élevé, le surplus de risque d’EIG par personne « vaccinée » est de : 0,583% – 0,513% = 0,06961433% (arrondi à 0,07%). La « vaccination » pour éviter une forme sévère de covid demeure défavorable, puisqu’elle entraîne encore 2710,75 x 0,06961433% = 1,89 EIG (calcul rapide :2500 x 0,07% = 1,75 EIG).
Selon les données de Pfizer, pour éviter une hospitalisation pour covid sévère, il était donc écrit que la campagne de vaccination en population générale (en moins bonne santé que dans l’essai) pouvait engendrer au moins 2 hospitalisations pour effet indésirable grave, et probablement bien davantage (ce chiffre pouvant être minoré par des événements graves n’ayant pas entraîné d’hospitalisation, passés inaperçus car non monitorés, et par les décès au domicile). Cette information non plus n’est jamais parvenue jusqu’à la population.
Conclusion
Depuis la fin des années quatre-vingt, nous savons que le principal facteur prédictif des conclusions des études cliniques ne se situe pas dans la méthodologie mais dans les liens d’intérêt entre le commanditaire ou le financeur de l’étude et ses cosignataires. L’essai clinique organisé par Pfizer, qui a scientifiquement fondé la « vaccination » obligatoire de tous les personnels et justifié l’exclusion des soignants réfractaires aux injections, en est une nouvelle illustration. En prenant au sérieux ses données et en suivant jusqu’au bout leur interprétation par les autorités, il était en effet écrit que pour éviter une hospitalisation pour covid grave, une campagne de vaccination en population générale entre 18 et 74 ans pouvait engendrer au moins deux hospitalisations pour effet indésirable grave, et probablement bien davantage.
Les autorités sanitaires comme les autorités gouvernementales des pays occidentaux, dont la France, n’ont pas communiqué aux citoyens ces éléments objectifs qui leur auraient permis de mesurer correctement les bénéfices et les risques du produit de Pfizer. Une communication institutionnelle biaisée et donc trompeuse a permis que ce rapport bénéfice-risque lourdement défavorable passe totalement inaperçu, d’où des décisions sanitaires individuelles et collectives largement inappropriées. Si cette carence des institutions publiques devait être mise en relation avec un rapport bénéfice-risque de la « vaccination » de la population générale d’emblée reconnu défavorable, la notification des résultats et les avis correspondants pourraient apparaître comme intentionnellement trompeurs, et la légitimité des décisions qui les ont suivis pourrait être remise en question.
Références
(1) Anglès A et al., Quels indicateurs d’efficacité des essais cliniques, pour quels résultats ? La Lettre du Pharmacologue, 16 (1), janvier-février 2002.
(2) Barnes DR, Bero LA, Why Review Articles on the Health Effects of Passive Smoking Reach Different Conclusions. JAMA, 279 (19), 20 mai 1998, 1566-70. doi: 10.1001/jama.279.19.1566
(3) Bobbio M, Demichelis B, Giustetto G. Completeness of reporting trial results : effect on physicians’ willingness to prescribe. Lancet, 343(8907), 14 mai 1994, 1209-11. Doi: 10.1016/S0140-6736(94)92407-4
(4) Boone MD et al., Pesticide Regulation amid the Influence of Industry. Bioscience, 64 (10), octobre 2014, 917-22. Doi: 10.1093/biosci/biu138
(5) Brown RB, Outcome Reporting Bias in COVID-19 mRNA Vaccine Clinical trials. Medicina, 57, 199, 26 février 2021. Doi: 10.3390/medicina57030199
(6) Communicating risks and benefits : an evidence-based user’s guide. Food and Drug Administration, FDA, US Department of Heath and Human Services. Silver Spring, MA, USA, 2011. https://www.fda.gov/about-fda/reports/communicating-risks-and-benefits-evidence-based-users-guide
(7) Cucchi M, Influence et pandémies, expériences hasardeuses et tentations autoritaires. Éditions Marco Pietteur, septembre 2022.
(8) Davidson R, Source of funding and outcome of clinical trials. Journal of General Internal Medicine, 1, 155–8, (1986). https://link.springer.com/article/10.1007/BF02602327
(9) Eschwège E, Bouvenot G, Doyon F, Lacroux A, Essais thérapeutiques mode d’emploi. Paris, Les Éditions INSERM & Le Quotidien du médecin, 2e édition, 1994.
(10) Frachon I, Médiator 150 mg – Combien de morts ? Brest, Éditions Dialogues, 2010.
(11) Guillemaud T, Lombaert E, Bourguet D, Conflicts of Interest in GM Bt Crop Efficacy and Durability Studies. PlosOne, 15 décembre 2016. Doi: 10.1371/journal.pone.0167777
(12) Herzberg N, Des effets indésirables fréquents a priori bénins. Le Monde, vendredi 20 décembre 2020, 9.
(13) Jouan A, Riché C, La santé en bande organisée. Paris. Éditions Robert Laffont, 2022.
(14) Kearns CE et al., Sugar Industry and Coronary Heart Disease Research A Historical Analysis of Internal Industry Documents Cardiology. JAMA Internal Medicine, 176 (11), novembre 2016, 1680-5. Doi: 10.1001/jamainternmed.2016.5394
(15) Lundh A et al., Industry sponsorship and research outcome. Cochrane Database of Systematic Reviews, 16 février 2017, 2. Doi: 10.1002/14651858.MR000033.pub3
(16) Million M, Dudouet P, Chabrière E, Cortaredona S, Brouqui P, Raoult D, Predictive factors of clinical assays during Covid-19. IHU Méditerranée Infection, avril 2020. Doi: 10.35088/aam9-bg48
(17) Million M, Dudouet P, Chabrière E, Cortaredona S, Roussel Y, Brouqui P, Raoult D, Predictive factors of clinical assays on hydroxychloroquine for COVID-19 mortality during the first year of the pandemic : a meta-synthesis. African Journal of Clinical and Environmental Microbiology, 31 janvier 2022, 23(1). Doi: 10.4314/ajcem.v23i1.1
(18) Polack FP et al., Safety and efficacy of the BNT162b2 mRNA covid-19 vaccine. New England Journal of Medicine, 31 décembre 2020, 383. Doi: 10.1056/NEJMoa2034577
(19) Robin M-M, Notre poison quotidien. Documentaire. Arte éditions/INA éditions, 2010.
(20) Vom Saal FS, An Extensive New Literature Concerning Low-Dose Effects of Bisphenol A Shows the Need for a New Risk Assessment. EHP, 1er août 2005. Doi: 10.1289/ehp.7713
Source : QG Média

Laisser un commentaire